![]()



Modular and user-friendly platform for AI-assisted rescoring of peptide identifications
About MS²Rescore
MS²Rescore performs ultra-sensitive peptide identification rescoring with LC-MS predictors such as MS²PIP and DeepLC, and with ML-driven rescoring engines Percolator or Mokapot. This results in more confident peptide identifications, which allows you to get more peptide IDs at the same false discovery rate (FDR) threshold, or to set a more stringent FDR threshold while still retaining a similar number of peptide IDs. MS²Rescore is ideal for challenging proteomics identification workflows, such as proteogenomics, metaproteomics, or immunopeptidomics.
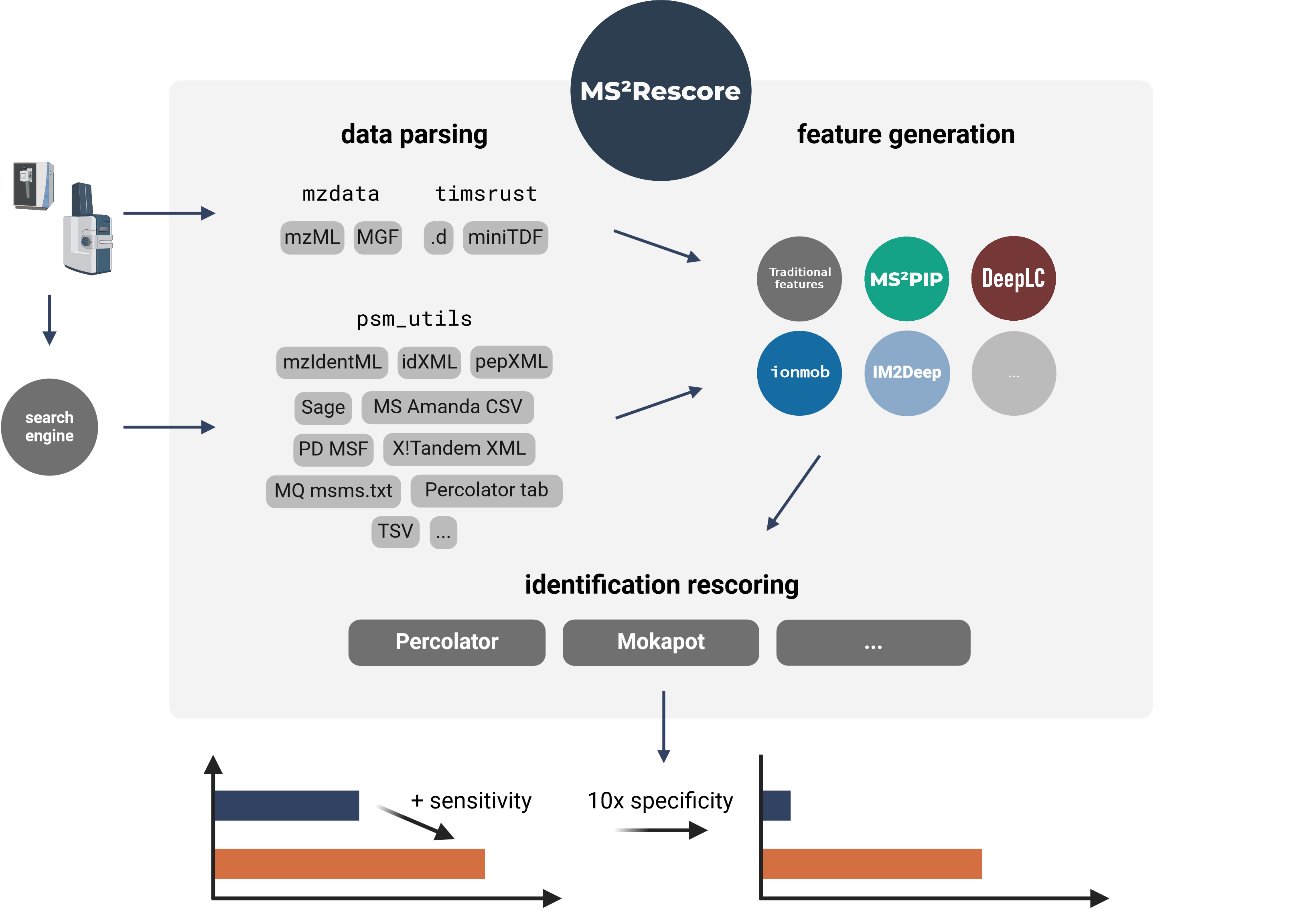
MS²Rescore can read peptide identifications in any format supported by psm_utils (see Supported file formats) and has been tested with various search engines output files:
- MS Amanda
.csv - Sage
.sage.tsv - PeptideShaker
.mzid - ProteomeDiscoverer
.msf - MSGFPlus
.mzid - Mascot
.mzid - MaxQuant
msms.txt - X!Tandem
.xml - PEAKS
.mzid
MS²Rescore is available as a desktop application, a command line tool, and a modular Python API.
TIMS²Rescore: Direct support for DDA-PASEF data
MS²Rescore v3.1+ includes TIMS²Rescore, a usage mode with specialized default configurations for DDA-PASEF data from timsTOF instruments. TIMS²Rescore makes use of new MS²PIP prediction models for timsTOF fragmentation and IM2Deep for ion mobility separation. Bruker .d and miniTDF spectrum files are directly supported through the timsrust library.
Checkout our paper for more information and the TIMS²Rescore documentation to get started.
Citing
Latest MS²Rescore publication:
MS²Rescore 3.0 is a modular, flexible, and user-friendly platform to boost peptide identifications, as showcased with MS Amanda 3.0. Louise Marie Buur, Arthur Declercq, Marina Strobl, Robbin Bouwmeester, Sven Degroeve, Lennart Martens, Viktoria Dorfer, and Ralf Gabriels. Journal of Proteome Research (2024) doi:10.1021/acs.jproteome.3c00785
*contributed equally
MS²Rescore for immunopeptidomics:
MS²Rescore: Data-driven rescoring dramatically boosts immunopeptide identification rates. Arthur Declercq, Robbin Bouwmeester, Aurélie Hirschler, Christine Carapito, Sven Degroeve, Lennart Martens, and Ralf Gabriels. Molecular & Cellular Proteomics (2021) doi:10.1016/j.mcpro.2022.100266
MS²Rescore for timsTOF DDA-PASEF data:
TIMS²Rescore: A DDA-PASEF optimized data-driven rescoring pipeline based on MS²Rescore. Arthur Declercq, Robbe Devreese, Jonas Scheid, Caroline Jachmann, Tim Van Den Bossche, Annica Preikschat, David Gomez-Zepeda, Jeewan Babu Rijal, Aurélie Hirschler, Jonathan R Krieger, Tharan Srikumar, George Rosenberger, Dennis Trede, Christine Carapito, Stefan Tenzer, Juliane S Walz, Sven Degroeve, Robbin Bouwmeester, Lennart Martens, and Ralf Gabriels. Journal of Proteome Research (2025) doi:10.1021/acs.jproteome.4c00609
Original publication describing the concept of rescoring with predicted spectra:
Accurate peptide fragmentation predictions allow data driven approaches to replace and improve upon proteomics search engine scoring functions. Ana S C Silva, Robbin Bouwmeester, Lennart Martens, and Sven Degroeve. Bioinformatics (2019) doi:10.1093/bioinformatics/btz383
To replicate the experiments described in this article, check out the publication branch of the repository.
Getting started
The desktop application can be installed on Windows with a one-click installer.
The Python package and command line interface can be installed with pip, conda, or docker.
Check out the full documentation to get started.
Questions or issues?
Have questions on how to apply MS²Rescore on your data? Or ran into issues while using MS²Rescore? Post your questions on the GitHub Discussions forum and we are happy to help!
How to contribute
Bugs, questions or suggestions? Feel free to post an issue in the issue tracker or to make a pull request!